Общая формула и строение молекул алкенов. Алкены: гомологический ряд, номенклатура и изомерия. Деление заместителей в бензольном кольце на два типа
Алкеновые углеводороды (олефины) являются одним из классов органических веществ, которым присущи свои . Виды изомерии алкенов у представителей данного класса не повторяются с изомерией других органических веществ.
Вконтакте
Характерные признаки класса
Этиленовыми олефинами именуют один из классов непредельных углеводородов, содержащих одну двойную связь.
По физическим свойствам представители данной категории непредельных соединений являются:
- газами,
- жидкостями,
- твердыми соединениями.
В составе молекул присутствует не только «сигма»-связь, но и «пи»-связь. Причиной этому является наличие в структурной формуле гибридизации «sp2 », которой свойственно расположение атомов соединения в одной плоскости.
При этом между ними формируется угол не менее ста двадцати градусов. Негибридизованным орбиталям «р » свойственно расположение как поверх молекулярной плоскости, так и под ней.
Такая особенность строения приводит к формированию дополнительных связей – «пи» или «π ».
Описанная связь менее прочна по сравнению с «сигма»-связями, так как перекрывание боком имеет слабое сцепление. Для суммарного распределения электронных плотностей образующихся связей характерна неоднородность. При вращении возле углерод-углеродной связи происходит нарушение перекрывания «р»-орбиталей. Для каждого алкена (олефина) такая закономерность является отличительным признаком.
Практически всем этиленовым соединениям присущи высокие температуры кипения и плавления, характерные не для всех органических веществ. Представители указанного класса непредельных углеводов быстро растворяются в и других растворителях органического состава.
Внимание! Ациклические непредельные соединения этиленовые углеводороды имеют общую формулу — C n H 2n.
Гомология
Исходя из того, что общая формула алкенов C n H 2n , им присуща определенная гомология. Гомологический ряд алкенов начинает первый представитель этилен или этен. Данное вещество в обычных условиях является газом и содержит два атома углерода и четыре атома водорода – C 2 H 4 . За этеном гомологический ряд алкенов продолжает пропен и бутен. Их формулы следующие: «C 3 H 6 » и «C 4 H 8 ». При обычных условиях они также являются газами, которые тяжелее , а значит, собирать их необходимо пробиркой, перевернутой вниз дном.
 Общая формула алкенов позволяет рассчитать следующего представителя данного класса, имеющего не менее пяти атомов углерода в структурной цепи. Это пентен с формулой «C 5 H 10 ».
Общая формула алкенов позволяет рассчитать следующего представителя данного класса, имеющего не менее пяти атомов углерода в структурной цепи. Это пентен с формулой «C 5 H 10 ».
По физическим характеристикам указанное вещество относится к жидкостям, так же как двенадцать следующих соединений гомологической линии.
Среди алкенов с указанными характеристиками есть и твердые вещества, которые начинаются с формулы C 18 H 36 . Жидким и твердым этиленовым углеводородам не свойственно растворение в воде, но при попадании в органические растворители они вступают с ними в реакцию.
Описанная общая формула алкенов подразумевает замену ранее стоявшего суффикса «ан» на «ен». Это закреплено правилами ИЮПАК. Какого бы представителя данной категории соединений мы не взяли, у них всех есть описанный суффикс.
В названии этиленовых соединений всегда присутствует определенная цифра, которая указывает на местоположение двойной связи в формуле. Примерами этого служит: «бутен-1» или «пентен-2». Атомную нумерацию начинают с того края, к которому ближе находится двойная конфигурация. Это правило является «железным» во всех случаях.
Изомерия
В зависимости от имеющегося вида гибридизации алкенов им присущи некоторые типы изомерии, каждый из которых имеет свои особенности и строение. Рассмотрим основные виды изомерии алкенов.
Структурного типа
Структурная изомерия подразделяется на изомеры по:
- углеродному скелету;
- расположению двойной связи.
Структурные изомеры углеродного скелета возникают в случае появления радикалов (ответвлений от главной цепи).
Изомерами алкенов указанной изомерии будут:
CH 2 =CH— CH 2 — CH 3.
2-метилпропен-1:
CH 2 =C— CH 3
│
У представленных соединений общее количество углеродных и водородных атомов (C 4 H 8), но разное строение углеводородного скелета. Это структурные изомеры, хотя свойства их не одинаковы. Бутену-1 (бутилену) присущ характерный запах и наркотические свойства, раздражающие дыхательные пути. Данными особенностями не обладает 2-метилпропен-1.
В данном случае нет изомеров у этилена (C 2 H 4), так как он состоит только из двух углеродных атомов, куда нельзя подставить радикалы.
Совет! Радикал разрешается ставить к средним и предпоследним углеродным атомам, но не разрешается располагать их около крайних заместителей. Данное правило работает для всех непредельных углеводородов.
Относительно расположения двойной связи различают изомеры:
CH 2 =CH— CH 2 — CH 2 -CH 3.
CH 3 -СH= CH— CH 2 -CH 3.
Общая формула алкенов у представленных примеров: C 5 H 10, , но местоположение одной двойной связи различное. Свойства указанных соединений будут различаться. Это структурная изомерия.

Изомерия
Пространственного типа
Пространственная изомерия алкенов связана с характером расположения углеводородных заместителей.
На основании этого различают изомеры:
- «Цис»;
- «Транс».
Общая формула алкенов позволяет создавать «транс-изомеры» и «цис-изомеры» у одного и того же соединения. Возьмем, к примеру, бутилен (бутен). Для него можно создать изомеры пространственного строения, по-разному расположив относительно двойной связи заместителей. С примерами изомерия алкенов будет выглядеть так:
«цис-изомер» «транс-изомер»

Бутен-2 Бутен-2
Из указанного примера видно, что у «цис-изомеров» по одну сторону плоскости расположения двойной связи находятся два одинаковых радикала. Для «транс-изомеров» это правило не работает, так как у них относительно углеродной цепи «С=С» располагаются два не похожих заместителя. Учитывая данную закономерность, можно самим строить «цис» и «транс» изомеры для различных ациклических этиленовых углеводородов.
Представленные «цис-изомер» и «транс-изомер» для бутена-2 невозможно превратить один в другой, так как для этого необходимо вращение вокруг имеющейся углеродной двойной цепочки (С=С). Чтобы осуществить данное вращение необходимо определенное количество энергии, чтобы разорвать существующую «p-связь».
На основании всего вышеизложенного можно сделать вывод, что изомеры «транс» и «цис» вида являются индивидуальными соединениями с определенным набором химических и физических свойств.
Нет изомеров у какого алкена. Пространственных изомеров не имеет этилен из-за одинакового расположения водородных заместителей относительно двойной цепи.
Межклассовые
Межклассовая изомерия у алкеновых углеводородов распространена значительно. Причиной этому служит сходность общей формулы представителей данного класса с формулой циклопарафинов (циклоалканов). У данных категорий веществ в одинаковое количество углеродных и водородных атомов, кратное составу (C n H 2n).
Межклассовые изомеры будут выглядеть так:
CH 2 =CH— CH 3.
Циклопропан:
Выходит, что формуле C 3 H 6 отвечают два соединения: пропен-1 и циклопропан. Из структурного строения видно разное расположение углерода относительно друг друга. По свойствам указанные соединения также разные. Пропен-1 (пропилен) – это газообразное соединение с низкой температурой кипения. Для циклопропана характерно газообразное состояние с резким запахом и едким вкусом. Химические свойства данных веществ также различаются, но состав у них идентичен. В органический данный вид изомеров именуют межклассовым.
Алкены. Изомерия алкенов. ЕГЭ. Органическая химия.
Алкены: Строение, номенклатура, изомерия
Вывод
Алкеновая изомерия – это их важная характеристика, благодаря которой в природе появляются новые соединения с другими свойствами, которые находят применение в промышленности и быту.
4. Химические свойства алкенов
Энергия двойной углерод-углеродной связи в этилене (146 ккал/моль) оказывается значительно более низкой, чем удвоенная энергия одинарной С-С-связи в этане (2 88=176 ккал/моль). -Связь С-С в этилене прочнее -связи, поэтому реакции алкенов, сопровождающиеся разрывом -связи с образованием двух новых простых -связей, представляют собой термодинамически благоприятный процесс. Так, например, в газовой фазе согласно расчетным данным все приведенные ниже реакции являются экзотермическими со значительной отрицательной энтальпией, независимо от их реального механизма.
С точки зрения теории молекулярных орбиталей также можно сделать вывод о большей реакционной способности -связи по сравнению с -связью. Рассмотрим молекулярные орбитали этилена (рис. 2).

Действительно, связывающая -орбиталь этилена имеет более высокую энергию, чем связывающая -орбиталь, и наоборот, разрыхляющая *-орбиталь этилена лежит ниже разрыхляющей *-орбитали связи С=С. В обычных условиях *- и *-орбитали этилена вакантны. Следовательно, граничными орбиталями этилена и других алкенов, определяющими их реакционную способность будут -орбитали.
4.1. Каталитическое гидрирование алкенов
Несмотря на то, что гидрирование этилена и других алкенов до алканов, сопровождается выделением тепла, эта реакция с заметной скоростью идет только в присутствии определенных катализаторов. Катализатор, по определению, не влияет на тепловой эффект реакции, и его роль сводится к понижению энергии активации. Следует различать гетерогенное и гомогенное каталитическое гидрирование алкенов. В гетерогенном гидрировании используются тонкоизмельченные металлические катализаторы - платина, палладий, рутений, родий, осмий и никель либо в чистом виде, либо нанесенные на инертные носители - BaSO 4 , CaCO 3 , активированный уголь, Al 2 O 3 и т. д. Все они нерастворимы в органических средах и действуют как гетерогенные катализаторы. Наибольшую активность среди них проявляют рутений и родий, но наибольшее распространение получил платина и никель. Платину обычно применяют в виде черного диоксида PtO 2 , широко известного под названием "катализатора Адамса". Диоксид платины получают при сплавлении платинохлористоводородной кислоты H 2 PtCl 6 . 6H 2 O или гексахлорплатината аммония (NH 4) 2 PtCl 6 с нитратом натрия. Гидрирование алкенов с катализатором Адамса проводят обычно при нормальном давлении и температуре 20-50 0 С в спирте, уксусной кислоте, этилацетате. При пропускании водорода двуокись платины восстанавливается непосредственно в реакционном сосуде до платиновой черни, которая и катализирует гидрирование. Другие более активные металлы платиновой группы используют на инертных носителях, например, Pd/C или Pd/BaSO 4 , Ru/Al 2 O 3 ; Rh/C и др. Палладий, нанесенный на уголь, катализирует гидрирование алкенов до алканов в спиртовом растворе при 0-20 0 С и нормальном давлении. Никель обычно используется в виде так называемого "никеля Ренея". Для получения этого катализатора сплав никеля с алюминием обрабатывают горячей водной щелочью для удаления почти всего алюминия и далее водой до нейтральной реакции. Катализатор имеет пористую структуру, и поэтому называется также скелетным никелевым катализатором. Типичные условия гидрирования алкенов над никелем Ренея требуют применения давления порядка 5-10 атм и температуры 50-100 0 С, т. е. этот катализатор значительно менее активен, чем металлы платиновой группы, но он белее дешев. Ниже приведены некоторые типичные примеры гетерогенного каталитического гидрирования ациклических и циклических алкенов:


Так как оба атома водорода присоединяются к атомам углерода двойной связи с поверхности металла-катализатора, обычно присоединение происходит с одной стороны двойной связи. Этот тип присоединения называется син -присоединением. В тех случаях когда два фрагмента реагента присоединяются с различных сторон кратной связи (двойной или тройной) имеет место анти -присоединение. Термины син - и анти - по смыслу эквивалентны терминам цис - и транс -. Для того, чтобы избежать путаницы и недоразумений термины син - и анти - относятся к типу присоединения, а термины цис - и транс - к строению субстрата.
Двойная связь в алкенах гидрируется с большей скоростью по сравнению со многими другими функциональными группами (С=О, COOR, CN и др.) и поэтому гидрирование двойной связи С=С часто представляет собой селективный процесс, если гидрирование ведется в мягких условиях (0-20 0 С и при атмосферном давлении). Ниже приведены некоторые типичные примеры:
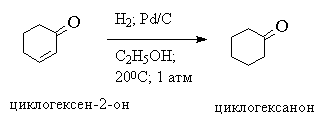

Бензольное кольцо не восстанавливается в этих условиях.
Большим и принципиально важным достижением в каталитическом гидрировании является открытие растворимых комплексов металлов, которые катализируют гидрирование в гомогенном растворе. Гетерогенное гидрирование на поверхности металлических катализаторов имеет ряд существенных недостатков, таких, как изомеризация алкенов и расщепление одинарных углерод-углеродных связей (гидрогенолиз). Гомогенное гидрирование лишено этих недостатков. За последние годы получена большая группа катализаторов гомогенного гидрирования - комплексов переходных металлов, содержащих различные лиганды. Лучшими катализаторами гомогенного гидрирования являются комплексы хлоридов родия (I) и рутения (III) с трифенилфосфином - трис(трифенилфосфин)родийхлорид (Ph 3 P) 3 RhCl (катализатор Уилкинсона) и гидрохлорид трис(трифенилфосфин)рутения (Ph 3 P) 3 RuHCl. Наиболее доступен родиевый комплекс, который получается при взаимодействии хлорида родия (III) с трифенилфосфином. Родиевый комплекс Уилкинсона используется для гидрирования двойной связи в обычных условиях.

Важное преимущество гомогенных катализаторов заключается в возможности селективного восстановления моно- или дизамещенной двойной связи в присутствии три- и тетразамещенной двойной связи из-за больших различий в скорости их гидрирования.

В случае гомогенных катализаторов присоединение водорода также происходит как син -присоединение. Так восстановление цис -бутена-2 дейтерием в этих условиях приводит к мезо -2,3-дидейтеробутану.

4.2. Восстановление двойной связи с помощью диимида
Восстановление алкенов до соответствующих алканов может быть с успехом осуществлено с помощью диимида NH=NH.
Диимид получают двумя основными методами: окислением гидразина пероксидом водорода в присутствии ионов Cu 2+ или взаимодействием гидразина с Ni-Ренея (дегидрирование гидразина). Если в реакционной смеси присутствует алкен, его двойная связь под действием очень нестабильного диимида подвергается гидрированию. Отличительной особенностью этого метода является строгая син -стереоспецифичность процесса восстановления. Полагают, что эта реакция протекает через циклический активированный комплекс со строгой ориентацией обеих реагирующих молекул в пространстве.

4.3. Реакции электрофильного присоединения по двойной связи алкенов
Граничными орбиталями ВЗМО и НСМО алкенов являются занятая - и пустая *-орбитали. Следовательно, в реакциях с электрофилами (Е +) будет участвовать -орбиталь, а в реакциях с нуклеофилами (Nu -) - *-орбиталь связи С=С (см. рис. 3). В большинстве случаев простые алкены легко вступают в реакции с электрофилами, а с нуклеофилами реагируют с большим трудом. Это объясняется тем, что обычно НСМО большинства электрофилов по энергии близки к энергии -ВЗМО алкенов, тогда как ВЗМО большинства нуклеофилов лежат значительно ниже *-НСМО.

Простые алкены реагируют лишь с очень сильными нуклеофильными агентами (карбанионы) в жестких условиях, однако введение электроноакцепторных групп в алкены, например, NO 2 , COR и др., приводит к понижению *-уровня, благодаря чему алкен приобретает способность реагировать с нуклеофилами средней силы (аммиак, RO - , Nє C - , енолят-анион и т. д.).
В результате взаимодействия электрофильного агента Е + с алкеном образуется карбокатион, обладающий высокой реакционной способностью. Карбокатион далее стабилизируется за счет быстрого присоединения нуклеофильного агента Nu - :


Поскольку медленной стадией является присоединение электрофила, то процесс присоединения любого полярного агента Е + Nu - следует рассматривать именно как электрофильное присоединение к кратной связи алкена. Известно большое число реакций этого типа, где роль электрофильного агента выполняют галогены, галогеноводороды, вода, соли двухвалентной ртути и другие полярные реагенты. Электрофильное присоединение к двойной связи в классификации механизмов органических реакций имеет символ Аd E (Addition Electrophilic ) и в зависимости от числа реагирующих молекул обозначается как Аd E 2 (бимолекулярная реакция) или Аd E 3 (тримолекулярная реакция).
4.3.а. Присоединение галогенов
Алкены реагируют с бромом и хлором с образованием продуктов присоединения по двойной связи одной молекулы галогена с выходом близким к количественному. Фтор слишком активен и вызывает деструкцию алкенов. Присоединение йода к алкенам в большинстве случаев представляет собой обратимую реакцию, равновесие которой смещено в сторону исходных реагентов.

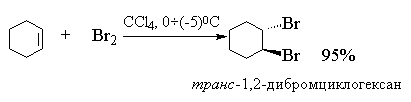

Быстрое обесцвечивание раствора брома в СCl 4 служит одним из простейших тестов на ненасыщенность, поскольку и алкены, и алкины, и диены быстро реагируют с бромом.
Присоединение брома и хлора к алкенам происходит по ионному, а не по радикальному механизму. Этот вывод следует из того, что скорость присоединения галогена не зависит от облучения, присутствия кислорода и других реагентов, инициирующих или ингибирующих радикальные процессы. На основании большого числа экспериментальных данных для этой реакции был предложен механизм, включающий несколько последовательных стадий. На первой стадии происходит поляризация молекулы галогена под действием электронов -связи. Атом галогена, приобретающий некоторый дробный положительный заряд, образует с электронами -связи нестабильный интермедиат, называемый -комплексом или комплексом с переносом заряда. Следует отметить, что в -комплексе галоген не образует направленной связи с каким-нибудь конкретным атомом углерода; в этом комплексе просто реализуется донорно-акцепторное взаимодействие электронной пары -связи как донора и галогена как акцептора.
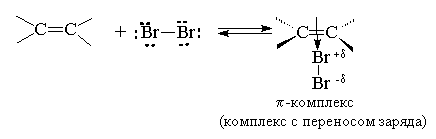
Далее -комплекс превращается в циклический бромониевый ион. В процессе образования этого циклического катиона происходит гетеролитический разрыв связи Br-Br и пустая р -орбиталь sp 2 -гибридизованного атома углерода перекрывается с р -орбиталью "неподеленной пары" электронов атома галогена, образуя циклический ион бромония.

На последней, третьей стадии анион брома как нуклеофильный агент атакует один из атомов углерода бромониевого иона. Нуклеофильная атака бромид-иона приводит к раскрытию трехчленного цикла и образованию вицинального дибромида (vic -рядом). Эту стадию формально можно рассматривать как нуклеофильное замещение S N 2 у атома углерода, где уходящей группой является Br+ .

Присоединение галогенов к двойной связи алкенов представляет собой одну из формально простых модельных реакций, на примере которой можно рассмотреть влияние основных факторов, позволяющих сделать аргументированные выводы о детальном механизме процесса. Для обоснованных выводов о механизме любой реакции следует располагать данными по: 1) кинетике реакции; 2) стереохимии (стереохимический результат реакции); 3) наличию или отсутствию сопряженного, конкурирующего процесса; 4) влиянию заместителей в исходном субстрате на скорость реакции; 5) использованию меченых субстратов и (или) реагентов; 6) возможности перегруппировок в ходе реакции; 7) влиянию растворителя на скорость реакции.
Рассмотрим эти факторы на примере галогенирования алкенов. Кинетические данные дают возможность установить порядок реакции по каждому компоненту и на этом основании сделать вывод об общей молекулярности реакции, т. е. о числе реагирующих молекул.
Для бромирования алкенов скорость реакции как правило описывается следующим уравнением:
v = k`[алкен] + k``[алкен] 2 ,
которое в редких случаях упрощается до
v = k`[алкен].
На основании кинетических данных можно сделать вывод о том, что в определяющей скорость стадии принимает участие одна или две молекулы брома. Второй порядок по брому означает, что с бромониевым ионом реагирует не бромид-ион Br - , а трибромид-ион , образующийся при взаимодействии брома и бромид-иона:
![]()
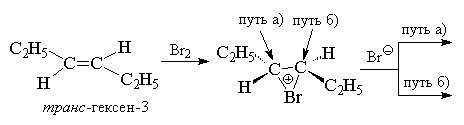
Это равновесие сдвинуто вправо. Кинетические данные не позволяют сделать какие-либо другие выводы о структуре переходного состояния и природе электрофильной частицы в реакции присоединения галогена по двойной связи. Наиболее ценную информацию о механизме этой реакции представляют данные по стереохимии присоединения. Присоединение галогена к двойной связи представляет собой стереоспецифический процесс (процесс, в котором образуется только один из возможных стереоизомеров; в стереоселективном процессе наблюдается преимущественное образование одного стереомера) анти -присоединения для алкенов и циклоалкенов, у которых двойная связь не сопряжена с бензольным кольцом. Для цис - и транс -изомеров бутена-2, пентена-2, гексена-3, циклогексена, циклопентена и других алкенов присоединение брома происходит исключительно как анти -присоединение. При этом в случае циклогексена образуется исключительно транс -1,2-дибромциклогексан (смесь энантиомеров).

Транс-расположение атомов брома в 1,2-дибромциклогексане можно упрощенно изобразить относительно средней плоскости циклогексанового кольца (без учета конформаций ):
При присоединении брома к циклогексену первоначально образуется транс -1,2-дибромциклогексан в а,а -конформации, которая затем сразу же переходит в энергетически более выгодную е,е -конформацию. Анти -присоединение галогенов к двойной связи позволяет отвергнуть механизм одностадийного синхронного присоединения одной молекулы галогена к двойной связи, которое может осуществляться только как син -присоединение. Анти -присоединение галогена не согласуется также и с образованием открытого карбкатиона RCH + -CH 2 Hal в качестве интермедиата. В открытом карбокатионе возможно свободное вращение вокруг С-С-связи, что должно приводить после атаки аниона Br - к образованию смеси продуктов как анти -, так и син -присоединения. Стереоспецифическое анти -присоединение галогенов явилось главной причиной создания концепции бромониевого или хлорониевого ионов в качестве дискретных промежуточных частиц. Эта концепция идеально удовлетворяет правилу анти -присоединения, поскольку нуклеофильная атака галогенид-иона возможна с анти -стороны по любому из двух атомов углерода галогенониевого иона по S N 2 механизму.

В случае несимметрично замещенных алкенов это должно приводить к двум энантиомерам трео -формы при присоединении брома к цис -изомеру или к энантиомерам эритро -формы при галоидировании транс -изомера. Это действительно наблюдается при присоединении брома, например, к цис - и транс -изомерам пентена-2.

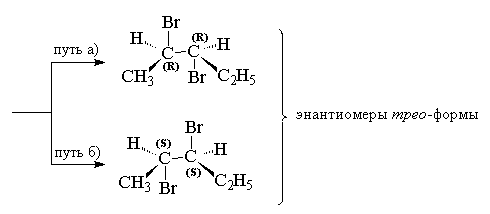


В случае бромирования симметричных алкенов, например, цис - или транс -гексенов-3 должны образоваться или рацемат (D,L -форма), или мезо -форма конечного дибромида, что и наблюдается в действительности.



Имеется независимое, прямое доказательство существования галогенониевых ионов в ненуклеофильной, индифферентной среде при низкой температуре. С помощью ЯМР-спектроскопии было зарегистрировано образование бромониевых ионов при ионизации 3-бром-2-метил-2-фторбутана при действии очень сильной кислоты Льюиса пятифтористой сурьмы в растворе жидкой двуокиси серы при -80 0 С.

Этот катион достаточно стабилен при -80 0 С в ненуклеофильной среде, но мгновенно разрушается при действии любых нуклеофильных агентов или при нагревании.
Циклические ионы бромония иногда могут быть выделены в чистом виде, если пространственные препятствия мешают их раскрытию при действии нуклеофилов:

Понятно, что возможность существования довольно стабильных в специальных условиях бромониевых ионов не может служить прямым доказательством их образования в реакции присоединения брома к двойной связи алкена в спирте, уксусной кислоте и других электронодонорных растворителях. Такие данные следует рассматривать лишь как независимое подтверждение принципиальной возможности образования галогенониевых ионов в процессе электрофильного присоединения по двойной связи.
Концепция галогенониевых иона позволяет дать рациональное объяснение обратимости присоединения йода к двойной связи. В катионе галогенония есть три электрофильных центра, доступных нуклеофильной атаке галогенид-аниона: два атома углерода и атом галогена. В случае хлорониевых ионов, анион Cl - , по-видимому, преимущественно или даже исключительно атакует углеродные центры катиона. Для бромониевого катиона равно вероятны оба направления раскрытия галогенониевого иона как за счет атаки бромид-иона по обоим атомам углерода, так и по атому брома. Нуклеофильная атака по атому брома бромониевого иона приводит к исходным реагентам брому и алкену:

Иодониевый ион раскрывается преимущественно в результате атаки иодид-иона по атому йода, и поэтому равновесие между исходными реагентами и иодониевым ионом смещено влево.

Кроме того, конечный продукт присоединения - вицинальный дииодид может подвергаться нуклеофильной атаке по атому йода присутствующим в растворе трииодид-анионом , что также приводит к образованию исходных реагентов алкена и иода. Другими словами, в условиях реакции присоединения происходит деиодирование образующегося вицинального дииодида под действием трииодид-аниона. Вицинальные дихлориды и дибромиды не дегалогенируются в условиях реакции присоединения соответственно хлора или брома к алкенам.
Анти-присоединение хлора или брома характерно для алкенов, у которых двойная связь не сопряжена с -электронами бензольного кольца. Для стирола, стильбена и их производных наряду с анти -присоединением имеет место и син -присоединение галогена, которое в полярной среде может стать даже доминирующим.

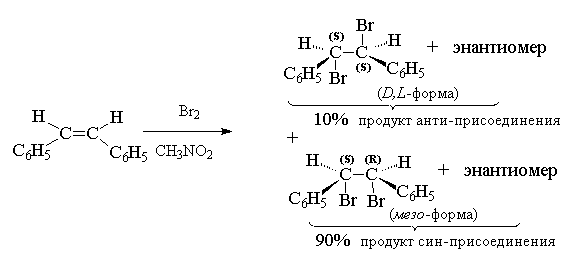
В тех случаях, когда присоединение галогена к двойной связи проводится в среде нуклеофильных растворителей, растворитель эффективно конкурирует с галогенид-ионом при раскрытии трехчленного цикла галогенониевого иона:

Образование продуктов присоединения с участием растворителя или какого-либо иного "внешнего" нуклеофильного агента носит название реакции сопряженного присоединения. При взаимодействии брома и стирола в метаноле образуется два продукта: вицинальный дибромид и бромэфир, соотношение которых зависит от концентрации брома в метаноле
В сильно разбавленном растворе доминирует продукт сопряженного присоединения, а в концентрированном растворе, напротив, преобладающий вицинальный дибромид. В водном растворе всегда преобладает галогенгидрин (спирт, содержащий галоген при -углеродном атоме) - продукт сопряженного присоединения.

ее-Конформер транс -2-хлорциклогексанола дополнительно стабилизирован водородной связью О-Н . . . Cl. В случае несимметричных алкенов в реакциях сопряженного присоединения галоген всегда присоединяется к атому углерода, содержащему наибольшее количество атомов водорода, а нуклеофильный агент к углероду с меньшим количеством атомов водорода. Изомерный продукт с иным расположением присоединяющихся групп не образуется. Это означает, что образующийся в качестве интермедиата циклический галогенониевый ион должен иметь несимметричную структуру с двумя различающимися по энергии и прочности связями С 1 -Hal и С 2 -Hal и большим положительным зарядом на внутреннем атоме углерода С 2 , что можно графически выразить двумя способами:

Поэтому нуклеофильной атаке растворителем подвергается атом углерода С 2 галогенониевого иона несмотря на то, что он более замещен и стерически менее доступен.


Один из лучших препаративных методов синтеза бромгидринов заключается в гидроксибромировании алкенов с помощью N-бромсукцинимида ( NBS ) в бинарной смеси диметилсульфоксида (ДМСО ) и воды.

Данную реакцию можно проводить в воде и без ДМСО , однако выходы бромгидринов в этом случае несколько ниже.



Образование продуктов сопряженного присоединения в реакции галогенирования алкенов также позволяет отвергнуть синхронный механизм присоединения одной молекулы галогена. Сопряженное присоединение к двойной связи находится в хорошем соответствии с двухстадийным механизмом с участием катиона галогенония в качестве интермедиата.
Для реакции электрофильного присоединения к двойной связи следует ожидать увеличения скорости реакции при наличии электронодонорных алкильных заместителей и ее уменьшения при наличии электроноакцепторных заместителей при двойной связи. Действительно, скорость присоединения хлора и брома к двойной связи резко возрастает при переходе от этилена к его метилзамещенным производным. Например, скорость присоединения брома к тетраметилэтилену в 10 5 раз выше, чем скорость его присоединения к бутену-1. Такое громадное ускорение определенно указывает на высокую полярность переходного состояния и высокую степень разделения зарядов в переходном состоянии и согласуется с элетрофильным механизмом присоединения.
В некоторых случаях присоединение хлора к алкенам, содержащим электронодонорные заместители, сопровождается отщеплением протона из промежуточного соединения вместо присоединения хлорид-иона. Отщепление протона приводит к образованию хлорзамещенного алкена, которое формально можно рассматривать как прямое замещение с миграцией двойной связи. Однако опыты с изотопной меткой указывают на более сложный характер происходящих здесь превращений. При хлорировании изобутилена при 0 0 С образуется 2-метил-3-хлорпропен (металлилхлорид) вместо ожидаемого дихлорида - продукта присоединения по двойной связи.

Формально как будто идет замещение, а не присоединение. Изучение этой реакции с использованием изобутилена меченного в положение 1 изотопом 14 С, показало, что прямое замещение водорода хлором не происходит, так как в образующемся металлилхлориде метка находится в группе 14 СН 2 Cl. Этот результат можно объяснить следующей последовательностью превращений:
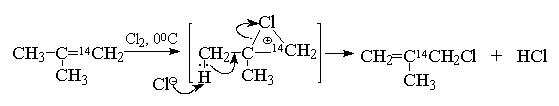
В отдельных случаях может происходить также 1,2-миграция алкильной группы

В ССl 4 (неполярный растворитель) эта реакция дает практически 100% дихлорида Б - продукта обычного присоединения по двойной связи (без перегруппировки).
Скелетные перегруппировки подобного типа наиболее характерны для процессов с участием открытых карбокатионов в качестве промежуточных частиц. Не исключено, что присоединение хлора в этих случаях идет не через хлорониевый ион, а через катионную частицу, близкую к открытому карбокатиону. Вместе с тем следует отметить, что скелетные перегруппировки явление достаточно редкое в процессах присоединения галогенов и смешанных галогенов по двойной связи: они чаще наблюдаются при присоединении хлора и гораздо реже при присоединении брома. Вероятность таких перегруппировок увеличивается при переходе от неполярных растворителей (ССl 4) к полярным (нитрометан, ацетонитрил).
Суммируя приведенные данные по стереохимии, сопряженному присоединению, влияние заместителей в алкене, а также перегруппировкам в реакциях присоединения галогенов по двойной связи, следует отметить, что они находятся в хорошем соответствии с механизмом электрофильного присоединения с участием циклического галогенониевого иона. Таким же образом могут быть интерпретированы данные по присоединению к алкенам смешанных галогенов, для которых стадийность присоединения определяется полярностью связи двух атомов галогена.
Алкены химически активны. Их химические свойства во многом определяются наличием двойной связи. Для алкенов наиболее характерны реакции электрофильного присоединения и реакции радикального присоединения. Реакции нуклеофильного присоединения обычно требуют наличие сильного нуклеофила и для алкенов не типичны. Алкены легко вступают в реакции окисления, присоединения а также способны к алильному радикальному замещению.
Реакции присоединения
Гидрирование Присоединение водорода (реакция гидрирования) к алкенам проводят в присутствии катализаторов. Чаще всего используют измельченные металлы - платину, никель, палладий и др. В результате образуются соответствующие алканы (насыщенные углеводороды).
$CH_2=CH_2 + H2 → CH_3–CH_3$
Присоединение галогенов. Алкены легко при обычных условиях вступают в реакции с хлором и бромом с образованием соответствующих дигалогеналканов, в которых атомы галогена находятся у соседних атомов углерода.
Замечание 1
При взаимодействии алкенов с бромом наблюдается обесцвечивание желто-бурой окраски брома. Это одна из старейших и самых простых качественных реакций на ненасыщенные углеводороды, поскольку аналогично реагируют также алкины и алкадиены.
$CH_2=CH_2 + Br_2 → CH_2Br–CH_2Br$
Присоединение галогеноводородов. При взаимодействии этиленовых углеводородов с галогеноводородами ($HCl$, $HBr$) образуются галогеналканы, направление реакции зависит от строения алкенов.
В случае этилена или симметричных алкенов реакция присоединения происходит однозначно и ведет к образованию только одного продукта:
$CH_2=CH_2 + HBr → CH_3–CH_2Br$
В случае несимметричных алкенов возможно образование двух разных продукта реакции присоединения:
Замечание 2
На самом деле в основном образуется только один продукт реакции. Закономерность направлении прохождения таких реакций установил российский химик В.В. Марковников в 1869 Она носит название правило Марковникова. При взаимодействии галогеноводородов с несимметричными алкенами атом водорода присоединяется по месту разрыва двойной связи в наиболее гидрированного атома углерода, то есть до того, что соединен с большим количеством атомов водорода.
Данное правило Марковников сформулировал на основе экспериментальных данных и только значительно позже оно получило теоретическое обоснование. Рассмотрим реакцию пропилена с хлористым водородом.
Одной из особенностей $p$-связи является его способность легко поляризоваться. Под влиянием метильной группы (положительный индуктивный эффект + $I$) в молекуле пропена электронная плотность $p$-связи смещается к одному из атомов углерода (= $CH_2$). Вследствие этого на нем возникает частичный отрицательный заряд ($\delta -$). На другом атоме углерода двойной связи в соответствии возникает частичный положительный заряд ($\delta +$).
Такое распределение электронной плотности в молекуле пропилена определяет место будущей атаки протоном. Это - атом углерода метиленовой группы (= $CH_2$), который несет частичный отрицательный заряд $\delta-$. А хлор, соответственно, атакует атом углерода с частичным положительным зарядом $\delta+$.
Как следствие, основным продуктом реакции пропилена с хлористым водородом является 2-хлорпропан.
Гидратация
Гидратация алкенов происходит в присутствии минеральных кислот и подчиняется правилу Марковникова. Продуктами реакции являются спирты
$CH_2=CH_2 + H_2O → CH_3–CH_2–OH$
Алкилирование
Присоединение алканов к алкенам в присутствии кислотного катализатора ($HF$ или $H_2SO_4$) при низких температурах приводит к образованию углеводородов с большей молекулярной массой и часто используется в промышленности для получения моторного топлива
$R–CH_2=CH_2 + R’–H → R–CH_2–CH_2–R’$
Реакции окисления
Окисление алкенов может происходить в зависимости от условий и видов окислительных реагентов как с разрывом двойной связи, так и с сохранением углеродного скелета:
Реакции полимеризации
Молекулы алкенов способны присоединяться при определенных условиях друг к другу с раскрытием $\pi$-связей и образования димеров, триммеров или высокомолекулярных соединений - полимеров. Полимеризация алкенов может протекать как по свободнорадикальному, так и катионно-анионому механизму. Как инициаторы полимеризации применяют кислоты, перекиси, металлы и др. Реакцию полимеризации осуществляют также под действием температуры, облучения, давления. Типичным примером является полимеризация этилена с образованием полиэтилена
$nCH_2=CH_2 → (–CH_2–CH_{2^–})_n$
Реакции замещения
Реакции замещения для алкенов не являются характерными. Однако при высоких температурах (свыше 400 ° C) реакции радикального присоединения, что носят обратимый характер, и подавляются. В этом случае становится возможным провести замещение атома водорода, находящегося в аллильном положении при сохранении двойной связи
$CH_2=CH–CH_3 + Cl_2 – CH_2=CH–CH_2Cl + HCl$
4.3.б. Присоединение галогеноводородов (гидрогалогенирование)
Другой важной реакций электрофильного присоединения к алкенам является давно известное гадрогалогенирование алкенов.
Ниже приведены типичные примеры присоединения HCl, HBr и HI к различным алкенам.


Влияние алкильных заместителей у двойной связи на скорость присоединения описывается следующей последовательностью:
R 2 C=CHR > RCH=CHR > RCH=CH 2
Это согласуется с таким механизмом, в котором в определяющей скорость стадии реакции происходит образование карбокатиона, поскольку стабильность алкильных катионов убывает в ряду третичный > вторичный > первичный. Таким образом, механизм присоединения должен включать промежуточное образование или свободного карбокатиона, что наблюдается редко, или интермедиата с карбокатионным характером. Последний случай наиболее типичен.
Если бы присоединение происходило через "свободный карбокатион", то реакция была бы совершенно нестереоселективной, так, как алкильные катионы имеют плоское строение. Однако, гидрогалогенирование, как правило, протекает стереоселективно, причем в зависимости от типа алкена может наблюдаться селективное анти -присоединение, селективное син - или смешанное син -анти -присоединение.
Для алкенов, у которых двойная связь не сопряжена с ароматическим кольцом, характерно анти -присоединение галогеноводорода. Анти -присоединение хлористого и бромистого водорода, хлористого и бромистого дейтерия наблюдается для циклогексена, циклопентена, 1,2-диметилгексена, 1,2-диметилпентена, цис - и транс -бутена-2, гексена-3 и многих других простых алкенов и циклоалкенов.
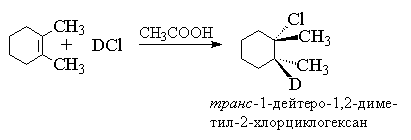
В продукте присоединения одинаковые заместители (метильные группы) расположены по разные стороны средней плоскости циклогексанового кольца, следовательно он относится к транс -ряду. Анти -присоединение трудно совместимо с механизмом, в котором предполагается образование дискретного карбокатиона. Для плоского карбокатиона нуклеофильная атака галогенид-иона равновероятна с обеих сторон плоскости, что должно привести к образованию смеси продуктов син - и анти -присоединения. Кинетика гидрогалогенирования алкенов также указывает на более сложный механизм присоединения. Для несопряженных алкенов скорость реакции описывается уравнением третьего порядка со вторым порядком по галогеноводороду, т. е. соответствует Ad E 3-механизму.
v = k [алкен] 2
Анти-присоединение и второй порядок реакции по галогеноводороду хорошо согласуется с Ad E 3-механизмом, в котором алкен взаимодействует с двумя молекулами галогеноводорода, одна из которых выполняет функцию электрофильного, а другая - нуклеофильного агента.

Такой тримолекулярный механизм предполагает, что первоначально образуется комплекс алкена и одной молекулой галогеноводорода с последующей атакой второй молекулы НХ на этот комплекс с анти -стороны без образования дискретного карбокатиона. Следует особо отметить, что любой тримолекулярный механизм должен состоять из двух последовательных стадий, поскольку одновременно столкновение трех молекул крайне маловероятно.

Анти-присоединение свидетельствует о предпочтительной нуклеофильной атаке галогеноводорода со стороны противоположной той, откуда происходит протонирование алкена. Вместо галогеноводорода функцию нуклеофильного агента в конечной стадии может выполнить и галогенид-ион. Действительно, скорость реакции обычно возрастает прямо пропорционально концентрации галогенид-иона, введенного в реакционную смесь в виде галогенидов тетраалкиламмония NR 4+ X - или лития LiX. В этом случае наблюдается стереоспецифическое анти -присоединение.
Для алканов, у которых двойная связь сопряжена с ароматическим кольцом, характерно син -присоединение или смешанное син -анти -присоединение галогеноводорода, например:

Син-присоединение является доминирующим процессом для цис - и транс -изомеров 1-фенилпропена, 1-фенил-4-алкилциклогексенов, аценафтилена, индена. При протонировании таких алкенов образуются карбокатионы бензильного типа, которые стабильнее чисто алкильных катионов, возникающих при протонировании обычных алкенов и циклоалкенов. Кинетика реакции в этом случае обычно описывается более простым уравнением второго порядка v = k[алкен], т. е. соответствует бимолекулярному Ad E 2-механизму. Ad E 2-Механизм предполагает образование ионной пары, включающей карбокатион и галогенид-ион.

Нельзя ожидать, что механизм присоединения с участием ионных пар будет отличаться высокой стереоселективностью. Если ионная пара превращается в конечный продукт быстрее, чем происходит вращение вокруг простой углерод-углеродной связи, конечным результатом будет син -присоединение, где протон и галогенид-ион присоединяется с одной и той же стороны двойной связи. В противном случае наблюдается образование продуктов как син - так и анти -присоединения НХ. Такой случай реализуется при гидрогалогенировании пара -замещенных стиролов Z-C 6 H 4 -CH=CH 2 . Наблюдаемая здесь закономерность заключается в том, что син -присоединение характерно лишь для тех олефинов, которые при протонировании дают относительно стабильный карбокатион, т. е. в случае донорных заместителей Z.
Для реакций гидрогалогенирования, протекающих по Ad E 2-механизму характерна конкуренция процессов сопряженного присоединения и перегруппировок, поскольку в качестве интермедиата образуется карбокатион или ионная пара.

В качестве примера перегруппировок с 1,2-миграцией алкильной группы и гидрид-иона приведем реакции гидрогалогенирования соответственно трет-бутилэтилена и изопропилэтилена.

При проведении этой же реакции без растворителя на холоду (-78 0 С) образуется смесь 33% нормального и 67% аномального (перегруппированного) продуктов присоединения.

4.3.в. Ориентация. Правило Марковникова
В отличие от симметричных электрофилов (Hal 2), галогеноводороды представляют собой несимметричные электрофильные реагенты. Присоединение любого несимметричного электрофила (HBr, ICl, H 2 O, Hg(OAc) 2 и т. д.) к несимметричному алкену в принципе могло бы дать смесь двух альтернативных продуктов, однако на практике обычно образуется только один из них. В качестве примера рассмотрим присоединение бромистого водорода к пропилену.

Еще в 1870 г. В.В. Марковников сформулировал эмпирическое правило, согласно которому несимметричные алкены присоединяют НХ таким путем, что преимущественно образуется продукт, в котором водород присоединяется к наименее замещенному, а Х - к наиболее замещенному концу двойной связи.
Обычно правило Марковникова объясняют различием в стабильности двух альтернативных карбокатионов. Например, в приведенном выше примере нормальный н -пропильный катион значительно менее стабилен, чем изопропильный катион, и поэтому реакция идет по второму пути.
Правило Марковникова первоначально использовалось только для случаев присоединения НХ к углеводородным субстратам, но в принципе его можно распространить и на реакции других замещенных алкенов. Так, присоединение НCl к CF 3 CH=CH 2 дает "анти -марковниковский" продукт CF 3 CH 2 CH 2 Cl. Этого и следовало ожидать, поскольку катион CF 3 CH+ CH 3 менее стабилен, чем катион CF 3 CH 2 CH 2+ из-за сильного (-I)-эффекта CF 3 -группы. Преимущественно образуется катион CF 3 CH 2 CH 2+ , но он тоже, хотя и в меньшей степени дестабилизирован индуктивным эффектом CF 3 -группы, вследствие чего присоединение HCl к трифторметилэтилену идет значительно медленнее, чем присоединение к незамещенному этилену.
По аналогичной причине катионы винилтриалкиламмония присоединяют HBr также против правила Марковникова:
Присоединение НХ к алкенам, имеющим сильные (-I) и (-M)-заместители, например, к акрилонитрилу или нитроэтилену также должно идти против правила Марковникова. Однако в этом случае двойная связь настолько сильно дезактивирована по отношению к электрофильным реагентам, что эти реакции идут лишь в очень жестких условиях. Хлористый винил СН 2 =СНСl всегда дает исключительно "марковниковские аддукты". Например, при его реакции с HCl образуется только 1,1-дихлорэтан (геминальный дихлорид) CH 3 CHCl 2 . Хлор, аналогично CF 3 -группе имеет сильный (-I)-эффект, и на первый взгляд, кажется, что по этой причине присоединение должно иметь антимарковниковскую ориентацию, т. к. катион + CH 2 CH 2 Cl должен быть более стабильным, чем катион СН 3 СН + Cl. Однако, в отличие от CF 3 -группы, хлор кроме (-I)-эффекта обладает также противодействующим ему (+М)-эффектом (т. к. имеет неподеленные пары). Опыт показывает, что величина мезомерного эффекта вполне достаточна, чтобы понизить энергию 1-хлорэтильного катиона ниже уровня энергии 2-хлорэтильного катиона, в котором +М-эффект не проявляется.

II. С позиций теории резонанса строение 1-хлорэтильного катиона может быть представлено следующим образом:
Тем не менее, присоединение к хлористому винилу происходит медленнее, чем к этилену в тех же условиях, т. е. по суммарному эффекту (-I > +M) хлор остается электроноакцепторным заместителем по сравнению с водородом, а 1-хлорэтильный катион менее стабилен, чем С 2 Н 5 + . Аналогичным образом реагируют с НХ и другие винилгалогениды.
Виниловые эфиры CH 2 =CHOR присоединяют НХ (X=Hal) по правилу Марковникова с гораздо большей скоростью, чем все перечисленные выше замещенные алкены. Это связано со значительным +М-эффектом RО-группы. В отличие от атома хлора, RО-группа по суммарному электронному эффекту (+М > -I) является сильным электронодонорным заместителем, эффективно стабилизирующим соседний карбокатионный центр. Строение карбокатиона в этом случае также может быть представлено в виде набора двух резонансных структур
Атака оксониевого катиона галогенид-анионом приводит к образованию -галогенэфиров типа СН 3 СН(Hal)OR.
4.3.г. Гидратация алкенов
Кислотно-катализируемая гидратация алкенов приводит к образованию спиртов. Направление гидратации алкенов определяется правилом Марковникова, поэтому предполагается, что в качестве промежуточной частицы в этой реакции образуется карбокатион.
Склонность вторичных алкильных карбокатионов к перегруппировкам мешает использованию гидратации алкенов для получения вторичных спиртов.

Этот метод в лаборатории нашел ограниченную область применения только для получения третичных спиртов. Реакция гидратации в этом случае в значительной степени обратима и третичные спирты образуются с низкими выходами (40-45%).
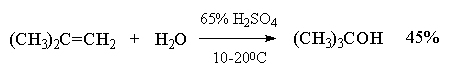

Гидратация простейших алкенов - этилена и пропилена - представляет собой важный промышленный метод получения этилового и изопропилового спиртов.

В лабораторной практике прямая гидратация алкенов не нашла широкого применения как вследствие жестких условий, так и благодаря образованию значительного количества изомерных спиртов. В настоящее время для региоселективного получения спиртов из алкенов обычно используется родственная реакция гидроксимеркурирования - демеркурирования.
4.3.д. Гидроксимеркурирование-демеркурирование
Электрофильная атака на двойную связь алкена может осуществляться ионами металлов, среди которых особое положение занимает катион ртути (II). Ацетат ртути в очень мягких условиях при 20 0 С присоединяется к алкенам в водном тетрагидрофуране (ТГФ) или в водной уксусной кислоте с образованием ртутьорганических соединений. Присоединение ацетата ртути по двойной связи протекает региоспецифично в строгом соответствии с правилом Марковникова, т. е. катион ртути присоединяется к наименее замещенному атому углерода.
Связь С-Hg в ртутьорганических соединениях может быть легко расщеплена под действием боргидрида натрия NaBH 4 , с образованием ртути и новой связи С-Н. Предполагается, что в качестве нестабильного интермедиата при этом получается алкилмеркургидрид, который далее разлагается с выделением металлической ртути по радикальному механизму.

Суммарно этот двухстадийный процесс гидроксимеркурирования-демеркурирования в конечном итоге представляет собой региоспецифичную гидратацию алкена по правилу Марковникова в исключительно мягких условиях, когда образование побочных продуктов сведено к предельно возможному минимуму. Это можно наглядно проиллюстрировать с помощью следующих примеров, в которых суммарный выход продуктов реакции составляет 90-98%. Приведенные цифровые данные в этом случае обозначают не выходы образующихся соединений, а их соотношение в смеси.




Как видно из приведенных выше примеров гидроксимеркурирование-демеркурирование алкенов в большинстве случаев обеспечивает региоспецифическую гидратацию алкенов с образованием практически только одного из двух изомерных спиртов. Следует отметить, что нет никакой необходимости в выделении ртутьорганического соединения, и оба процесса могут быть проведены непосредственно один за другим.
Гидроксимеркурирование несимметричных алкенов, по-видимому, начинается с атаки катиона AcOHg + и образования в качестве интермедиата несимметричного циклического меркуриниевого катиона (аналога несимметричного галогенониевого иона), который затем раскрывается в результате нуклеофильной атаки водой по наиболее замещенному атому углерода, несущему больший положительный заряд.
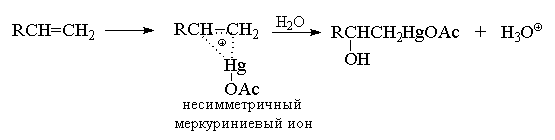
Мостиковый меркуриниевый ион можно зафиксировать в ненуклеофильной сильнокислой среде даже при 20 0 С при присоединении более сильного электрофильного агента - трифторацетата ртути в смеси фторсульфоновой кислоты и пятифтористой сурьмы.

Меркуриниевый катион может расщепляться не только при действии воды, но и других электронодонорных растворителей: спиртов, уксусной кислоты, ацетонитрила и др. Конечным продуктом реакции в этом случае будут соответственно простые эфиры, ацетаты или N-замещенные амиды уксусной кислоты, например:


При использовании в реакции алкоксимеркурирования-демеркурирования разветвленных вторичных или третичных спиртов более эффективными, чем ацетат ртути, являются трифторацетат Hg(OCOCF 3) 2 или трифлат ртути Hg(OSO 2 CF 3) 2 .

Таким образом, гидрокси- и алкоксимеркурирование-демеркурирование - это один из лучших препаративных методов синтеза спиртов и простых эфиров с разветвленными алкильными радикалами.
Присоединение солей ртути к алкенам представляет собой наиболее яркий пример реакции сопряженного присоединения к двойной связи, где роль внешнего нуклеофильного агента выполняет растворитель. Стереохимия двойного процесса гидроксимеркурирования-демеркурирования зависит от стереохимического результата каждой отдельной стадии. Для гидроксимеркурирования характерно анти -присоединение, как и для других реакций с участием циклического катиона. Однако радикальное демеркурирование не отличается высокой стереоселективностью. Поэтому весь процесс в целом также нестереоспецифичен.
4.3.е. Присоединение алкенов (катионная димеризация и полимеризация алкенов)
Наиболее интересным примером такого типа реакций является димеризация и полимеризация изобутилена в присутствии серной кислоты.

Техническое название этой смеси алкенов - "диизобутилен". Данная реакция протекает по катионному механизму, сходному с механизмом присоединения минеральных кислот к двойной связи алкенов. На первой стадии протон присоединяется к молекуле изобутилена с образованием относительно стабильного трет -бутилкатиона. Далее образовавшийся трет -бутилкатион (кислота Льюиса) реагирует с молекулой изобутилена (основание Льюиса) с образованием нового стабильного третичного октильного катиона.

В данных условиях под действием оснований (H 2 O, HSO 4 - -ионы) октильный карбокатион быстро теряет протон и превращается в смесь изомерных пентенов, т. к. отрыв протона происходит из двух различных положений:

Преимущественное образование термодинамически менее стабильного алкена - 2,4,4-триметилпентена-1 (80% в реакционной смеси) связано с большей пространственной доступностью для атаки основанием атомов водорода метильных групп, по сравнению с атомами водорода метиленовой группы. В промышленности "диизобутилен" гидрируют на Ni-Ренея и получают "изооктан" (техническое название углеводорода 2,2,4-триметилпентана), используемый в качестве высокооктанового топлива для двигателей внутреннего сгорания.
При высоких концентрациях серной кислоты (более 80%) происходит катионная полимеризация изобутилена с образованием полимера, называемого полиизобутиленом (-CH 2 C(CH 3) 2 -) n . Этот каучукоподобный полимер используют для получения антикоррозионных и гидроизоляционных покрытий, герметиков и др.
Кроме изобутилена, по катионному механизму полимеризуются 3-метилбутен-1, виниловые эфиры и некоторые производные стирола, способные образовывать сравнительно устойчивые карбокатионы. В качестве катализаторов катионной полимеризации также используют фтористый водород и кислоты Льюиса: BF 3 , AlCl 3 , AlBr 3 и др. в присутствии очень малых количеств воды.
4.3.ж. Присоединение алканов (алкилирование алкенов)
Другой промышленный метод синтеза "изооктана" основан на взаимодействии изобутилена с избытком изобутана в присутствии концентрированной серной кислоты или в безводном фтористом водороде при 0 10 0 С.

Эта реакция также протекает по катионному механизму и, что особенно интересно, является примером цепного катионного процесса. Сначала изобутилен в условиях реакции димеризуется с образованием третичного октильного катиона (CH 3) 3 CCH 2 C + (CH 3) 2 . Подробно механизм его образования изложен в предыдущем разделе. Далее происходит быстрый перенос водорода (в виде гидрид-иона) от изобутана к октильному катиону с образованием молекулы "изооктана" и нового трет-бутилкатиона, который в свою очередь быстро реагирует с изобутиленом с образованием нового октильного катиона и т. д.

Кроме синтеза "изооктана", такой метод алкилирования используется в нефтехимической промышленности для синтеза высококипящих разветвленных углеводородов из разветвленных алкенов и алканов низкокипящих фракцией термического крекинга.
К непредельным относят углеводороды, содержащие в молекулах кратные связи между атомами углерода. Непредельными являются алкены, алкины, алкадиены (полиены) . Непредельным характером обладают также циклические углеводороды, содержащие двойную связь в цикле (циклоалкены ), а также циклоалканы с небольшим числом атомов углерода в цикле (три или четыре атома). Свойство «непредельности» связано со способностью этих веществ вступать в реакции присоединения, прежде всего водорода, с образованием предельных, или насыщенных углеводородов - алканов.
Строение алкенов
Ациклические углеводороды, содержащие в молекуле помимо одинарных связей, одну двойную связь между атомами углерода и соответствующие общей формуле СnН2n. Свое второе название - олефины
- алкены получили по аналогии с жирными непредельными кислотами (олеиновая, линолевая), остатки которых входят в состав жидких жиров - масел.
Атомы углерода, между которыми есть двойная связь, находятся в состоянии sр 2 -гибридизации. Это означает, что в гибридизации участвуют одна s- и две р-орбитали, а одна р-орбиталь остается негибридизованной. Перекрывание гибридных орбиталей приводит к образованию σ-связи, а за счет негибридизованных р-орбиталей
соседних атомов углерода образуется вторая, π-связь. Таким образом, двойная связь состоит из одной σ- и одной π — связи. Гибридные орбитали атомов, образующих двойную связь, находятся в одной плоскости, а орбитали, образующие π -связь, располагаются перпендикулярно плоскости молекулы. Двойная связь (0,132 им) короче одинарной, а ее энергия больше, т. к. она является более прочной. Тем не менее, наличие подвижной, легко поляризуемой π -связи приводит к тому, что алкены химически более активны, чем алканы, и способны вступать в реакции присоединения.

Строение этилена

Образование двойной связи в алкенах
Гомологический ряд этена
Неразветвленные алкены составляют гомологи- ческий ряд этена (этилена ): С 2 Н 4 - этен, С 3 Н 6 - пропен, С 4 Н 8 - бутен, С 5 Н 10 - пентен, С 6 Н 12 - гексен, С 7 Н 14 - гептен и т.д.
Изомерия алкенов
Для алкенов характерна структурная изомерия. Структурные изомеры отличаются друг от друга строением углеродного скелета. Простейший алкен, для которого характерны структурные изомеры, - это бутен:

Особым видом структурной изомерии является изомерия положения двойной связи:
Алкены изомерны циклоалканам (межклассовая изомерия), например:

Вокруг одинарной углерод-углеродной связи возможно практически свободное вращение атомов углерода, поэтому молекулы алканов могут приобретать самую разнообразную форму. Вращение вокруг двойной связи невозможно, что приводит к появлению у алкенов еще одного вида изомерии - геометрической, или цис- и транс- изомерии .


Цис-изомеры
отличаются от транс-изомеров
пространственным расположением фрагментов молекулы (в данном случае метильных групп) относительно плоскости π -связи, а следовательно, и свойствами.
Номенклатура алкенов
1. Выбор главной цепи.
Образование названия углеводорода начинается с определения главной цепи - самой длинной цепочки атомов углерода в молекуле. В случае алкенов главная цепь должна содержать двойную связь.
2. Нумерация атомов главной цепи.
Нумерация атомов главной цепи начинается с того конца, к которому ближе находится двойная связь.
Например,правильное название соединения:
![]()
Если по положению двойной связи нельзя определить начало нумерации атомов в цепи, то его определяет положение заместителей так же, как для предельных углеводородов.

3. Формирование названия. В конце названия указывают номер атома углерода, у которого начинается двойная связь, и суффикс -ен , обозначающий принадлежность соединения к классу алкенов. Например:

Физические свойства алкенов
Первые три представителя гомологического ряда алкенов - газы; вещества состава С5Н10 — С16Н32 - жидкости; высшие алкены - твердые вещества.
Температуры кипения и плавления закономерно повышаются при увеличении молекулярной массы соединений.
Химические свойства алкенов
Реакции присоединения
. Напомним, что отличительной чертой представителей непредельных углеводородов - алкенов является способность вступать в реакции присоединения. Большинство этих реакций протекает по механизму электрофильного присоединения
.
1. Гидрирование алкенов.
Алкены способны присоединять водород в присутствии катализаторов гидрирования, металлов - платины, палладия, никеля:
Эта реакция протекает при атмосферном и повышенном давлении и не требует высокой температуры, т. к. является экзотермической. При повышении температуры на тех же катализаторах может пойти обратная реакция - дегидрирование.
2. Галогенирование (присоединение галогенов)
. Взаимодействие алкена с бромной водой или раствором брома в органическом растворителе (СС14) приводит к быстрому обесцвечиванию этих растворов в результате присоединения молекулы галогена к алкену и образования дигалогеналканов.
3. Гидрогалогенирование (присоединение галогеноводорода)
.
Эта реакция подчиняется
При присоединении галогеноводорода к алкену водород присоединяется к более гидрированному атому углерода, т. е. атому, при котором находится больше атомов водорода, а галоген - к менее гидрированному.


4. Гидратация (присоединение воды).
Гидратация алкенов приводит к образованию спиртов. Например, присоединение воды к этену лежит в основе одного из промышленных способов получения этилового спирта.
Обратите внимание на то, что первичный спирт (с гидроксогруппой при первичном углероде) образуется только при гидратации этена. При гидратации пропена или других алкенов образуются вторичные спирты .

Эта реакция протекает также в соответствии с правилом Марковникова - катион водорода присоединяется к более гидрированному атому углерода, а гидроксогруппа - к менее гидрированному.
5. Полимеризация.
Особым случаем присоединения является реакция полимеризации алкенов:
Эта реакция присоединения протекает по свободнорадикальному механизму.
Реакции окисления.
1. Горение.
Как и любые органические соединения, алкены горят в кислороде с образованием СО2 и Н2О:
2. Окисление в растворах. В отличие от алканов алкены легко окисляются под действием растворов перманганата калия. В нейтральных или щелочных растворах происходит окисление алкенов до диолов (двухатомных спиртов), причем гидроксильные группы присоединяются к тем атомам, между которыми до окисления существовала двойная связь:



